How to Prepare DNA Libraries for NGS Efficiently?
The field of genomics is rapidly evolving, and efficient DNA Library Preparation for NGS plays a crucial role. Dr. Emily Chen, a leading expert in the field, once emphasized, "Optimizing library preparation can significantly impact sequencing results." Her insight highlights the importance of getting it right from the start.
Effective library preparation is not a one-size-fits-all process. It requires careful consideration of various factors, including sample quality and desired throughput. In many labs, preparation can become a bottleneck. Common pitfalls include inadequate quality control and overlooking the importance of proper protocols. These missteps can lead to suboptimal data.
As we strive for precision, it's essential to reflect on our methods continuously. Each step in the DNA Library Preparation for NGS needs scrutiny, especially when faced with new technologies. Embracing a mindset of improvement can help mitigate potential errors and enhance the overall efficiency of the process.

Understanding the Basics of DNA Library Preparation for NGS
Preparing DNA libraries for next-generation sequencing (NGS) involves several critical steps. The process begins with DNA extraction from samples. Quality matters here. You want intact DNA. A spectrophotometer can help assess concentration and purity. Often, samples come with varying quality. Some may require additional purification steps, which can be frustrating.
Following extraction, fragmentation is needed. This breaks DNA into manageable sizes. Different methods exist, like enzymatic or mechanical shearing. Each method has pros and cons. Enzymatic methods tend to be gentler. However, they can be costly and time-consuming. Mechanical methods are faster but may introduce bias. Balancing these factors is crucial for efficient preparation.
Next comes adaptor ligation. Adaptor sequences are crucial for sequencing, but they can be tricky to work with. Improper ligation can lead to low yield. This step requires precision. A common issue is the inefficient attachment of adaptors, which leads to wasted time. Finally, PCR amplification enriches the library. However, over-amplification can bias results. Each step must be optimized based on sample type and desired outcome. Such careful adjustments can significantly enhance library quality, but they require a lot of trial and error.
NGS DNA Library Preparation Steps
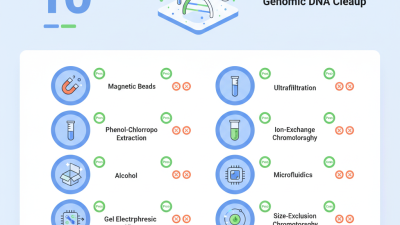
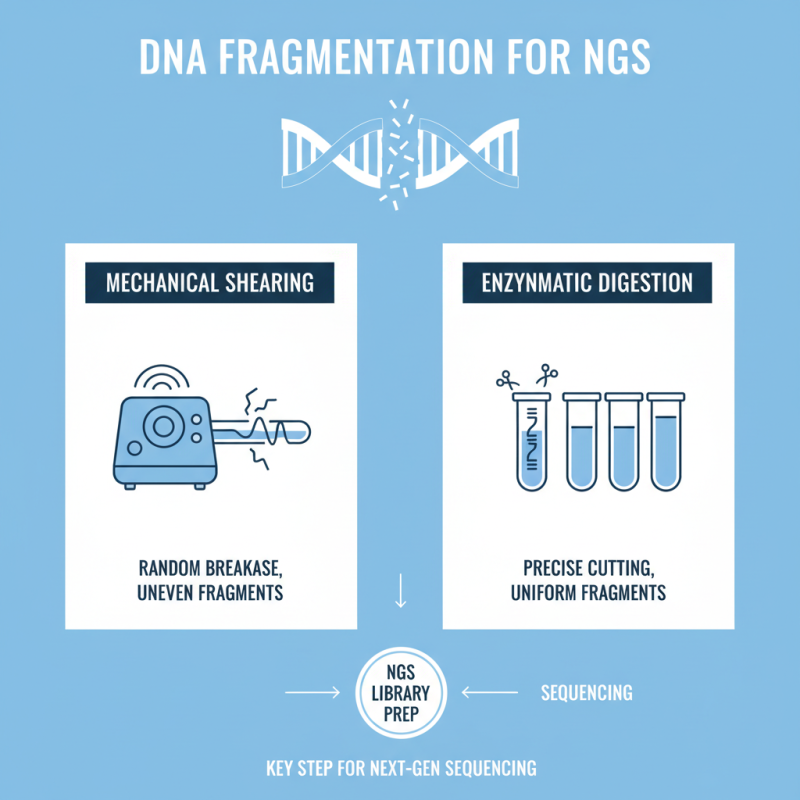
Key Steps in DNA Fragmentation and Size Selection
DNA fragmentation is a critical step in preparing libraries for next-generation sequencing (NGS). This process breaks the DNA into smaller, manageable pieces. Various methods can achieve this, such as mechanical shearing or enzymatic digestion. Each method has its own advantages and disadvantages. Mechanical shearing may produce uneven fragment sizes. On the other hand, enzymatic digestion can lead to more uniform fragments, but it requires careful optimization.
After fragmentation, size selection is crucial. This step ensures that only DNA fragments of the desired size range are used for library construction. A common approach is gel electrophoresis. However, running gels takes time and can be challenging. An alternative method is using magnetic beads. They allow for rapid size selection, but careful handling is necessary to avoid loss of sample. It's easy to overlook small fragments, which might be essential for your analysis.
In any case, it’s important to assess the quality of the fragments after size selection. Rotating between different methods may yield better results. Finding the right balance requires experimentation and adjustment. Mistakes can happen, leading to poor quality libraries. Regularly reviewing your processes can help refine your technique. There is always room for improvement in DNA library preparation for NGS.
Library Amplification: Techniques and Best Practices
Library amplification is a crucial step in preparing DNA libraries for next-generation sequencing (NGS). This process boosts the quantity of DNA fragments, ensuring sufficient material for sequencing. One effective technique for amplification is PCR. It selectively replicates target regions. However, PCR can introduce biases. Some fragments may amplify more than others. This uneven amplification can affect data quality.
Another promising approach is the use of enzymatic amplification. Unlike PCR, this method can reduce biases. It involves using specific enzymes to replicate DNA fragments evenly. This technique often enhances the representation of GC-rich regions. Nevertheless, some researchers find it less straightforward than PCR. Understanding the optimal conditions can be challenging.
Best practices for library amplification include monitoring reaction conditions closely. Adjusting temperature and enzyme concentrations can make a significant difference. Regularly checking fragment sizes is also important. If unexpected sizes appear, it may indicate issues. Documenting every step helps in troubleshooting later. Amplification requires attention and precision. Mistakes can lead to insufficient library yield or poor sequencing results.
Efficient Quantification and Quality Control of DNA Libraries
Efficient quantification and quality control of DNA libraries are crucial steps in preparing for next-generation sequencing (NGS). Accurate quantification ensures that the right amount of DNA is used for sequencing. Too much DNA can lead to excess noise, while too little may result in inadequate coverage. Various methods exist for quantification, including fluorometric assays, spectrophotometry, and qPCR. Each method has its pros and cons. It's essential to choose the one that fits your needs.
The quality control aspect often involves checking for size distribution and purity. Techniques like agarose gel electrophoresis and Bioanalyzer traces provide valuable insights. These methods can reveal whether the DNA is intact and if there are any contaminants present. However, user error can affect results. Relying on a single technique may not provide a complete picture. Balancing efficiency with thoroughness is key. Regularly reviewing protocols and validating methods can improve reliability. This iterative process ensures that your DNA libraries are of high quality and ready for successful NGS.
How to Prepare DNA Libraries for NGS Efficiently? - Efficient Quantification and Quality Control of DNA Libraries
| Sample ID |
DNA Concentration (ng/μL) |
Quality (A260/A280) |
Fragment Size (bp) |
Binding Efficiency (%) |
| Sample 1 |
50 |
1.8 |
300 |
85 |
| Sample 2 |
120 |
1.9 |
200 |
90 |
| Sample 3 |
75 |
1.6 |
150 |
80 |
| Sample 4 |
30 |
2.0 |
500 |
70 |
| Sample 5 |
100 |
1.7 |
400 |
88 |
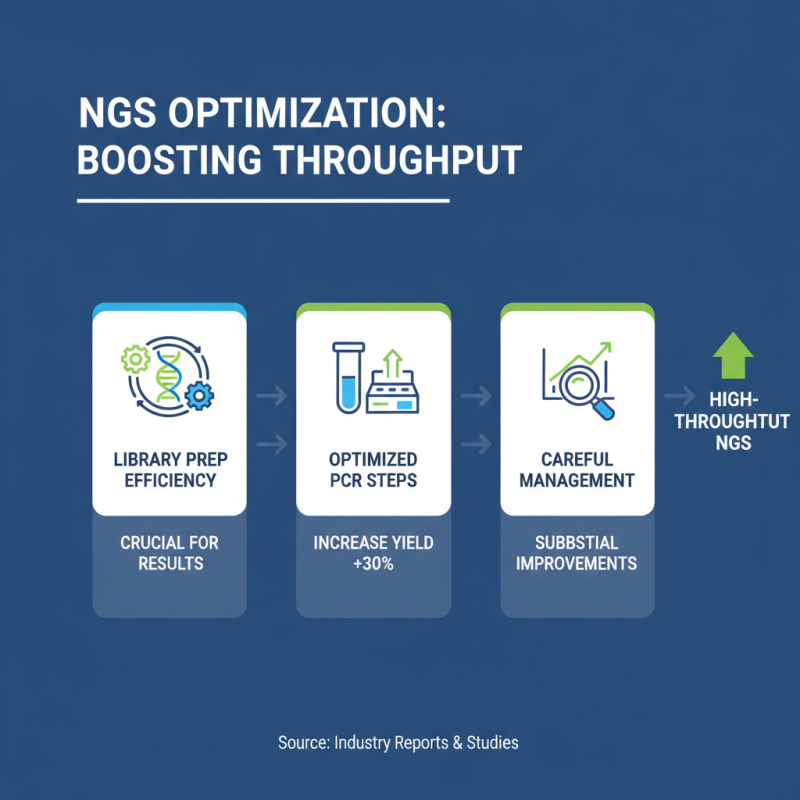
Optimizing Sequencing for High-Throughput Results in NGS
Optimizing sequencing for high-throughput results in next-generation sequencing (NGS) is crucial. According to recent industry reports, the efficiency of library preparation can significantly impact overall results. Studies indicate that optimizing the PCR steps can increase yield by up to 30%. This suggests that careful management of conditions can lead to substantial improvements.
Another point of consideration is the choice of index sequences. Incorrect indexing can lead to erroneous data. A report from a major sequencing consortium highlighted that about 15% of reads mapped incorrectly due to indexing failures. This emphasizes the need for rigorous quality controls in the library preparation stage.
Additionally, evaluating the average fragment size can be revealing. Research shows that libraries with consistently sized fragments typically yield more reliable data. However, achieving uniform fragment sizes can be challenging. Variability in sample input can lead to a mixed output. Hence, optimizing the sample preparation process is essential, even though it often requires multiple trial runs. Striving for perfection in these processes is important, but it is equally vital to learn from these challenges.